Глава 6. Методы
определения антиокислительной активности
6.1. Общая характеристика системы
антиоксидантной защиты организма
Образование
АФК и окислительная модификация важнейших биомолекул свойственно нормально метаболизирующей
клетке. Окисление идет со значительной скоростью, но стационарная концентрация
продуктов пероксидации довольно мала вследствие
наличия сложной системы взаимодействующих путей ее регуляции.
Принято
делить химические соединения и физические воздействия, влияющие на скорость
перекисного окисления липидов, на прооксиданты
(усиливают свободнорадикальные процессы) и
антиоксиданты (тормозят свободнорадикальные
процессы). К прооксидантам в живой клетке относятся
высокие концентрации кислорода (например, при длительной гипербарической оксигенации больного), ферментные системы, генерирующие супероксидные радикалы (например, ксантиноксидаза,
ферменты плазматической мембраны фагоцитов и др.), ионы металлов переменной
валентности (железо, медь).
Под действием тех или иных агентов
скорость ПОЛ может изменяться, однако системы, регулирующие ПОЛ, обладают
способностью быстро возвращать уровень ПОЛ к норме. Организм обладает
многоуровневой стратегией защиты от повреждающего действия АФК. Защита
осуществляется двумя принципиально различными механизмами:
1) снижением образования первой АФК –
О2![]() путем уменьшения О2 в клетке или его более
быстрого использования дыхательной цепью ввиду снятия ее контроля ∆µН+;
путем уменьшения О2 в клетке или его более
быстрого использования дыхательной цепью ввиду снятия ее контроля ∆µН+;
2) функционированием специальной АОС,
выработанной в процессе эволюции аэробных организмов.
Основными функциями этой системы являются: ограничение интенсивности реакций свободнорадикального и перекисного окисления; защита
чувствительных к окислительным повреждениям биомолекул
мембран, внутри- и внеклеточных структур от действия свободных радикалов и
перекисных соединений; восстановление окислительных молекулярных повреждений.
Существуют
различные классификации компонентов антиоксидантной системы. Так, по механизмам
действий в АОС выделяют антирадикальную (первичную) и
антиперекисную (вторичную) защиту. По молекулярному
строению среди антиоксидантов выделяют ферменты и неферментные соединения,
которые, в свою очередь, могут быть высоко- и низкомолекулярными. Часть
защитных систем клетки локализуется в липидной фазе, а часть – в водной фазе.
По отношению к действию в водной или липидной фазе антиоксиданты делят на водо-
и жирорастворимые. По локализации действия относительно клеточных структур
антиоксиданты разделяют на внутриклеточные, мембранные
и экстрацеллюлярные. Кроме того, по происхождению
антиоксиданты делят на биооксиданты, природные и
синтетические соединения. В табл. 4 приведены наиболее известные антиоксиданты.
Таблица 4. Антиоксиданты в живых
системах
Антиоксиданты |
Локализация |
Функция |
|
Ферменты |
||
|
Cu, Zn-СОД |
Эритроциты, цитоплазма, межмембранное пространство митохондрий |
Тушение О2 |
|
Mn-СОД |
Митохондрии |
Тушение О2 |
|
Внеклеточная СОД |
Плазма крови, стенки сосудов Пероксисомы |
Тушение О2 |
|
Глутатионпероксидаза |
Цитоплазма, митохондрии |
Тушение Н2О2 Деградация Н2О2
и перекисей липидов |
|
Глутатионтрансфераза |
Клеточные
мембраны, митохондрии, эндоплазматический ретикулум, цитозоль |
Деградация перекисей
липидов |
|
Глутатионредуктаза |
Цитоплазма |
Восстанавливает
окисленный глутатион |
|
Белки |
||
|
Ферритин |
Цитоплазма |
Хелатор Fe2+ |
|
Трансферрин |
Внеклеточная среда |
Хелатор Fe2+ |
|
Лактоферрин |
Внеклеточная среда |
Хелатор Fe2+ |
|
Церулоплазмин |
Внеклеточная среда |
Хелатор Cu2+, окисление Fe2+, тушение О2 |
|
Альбумин |
Внеклеточная среда |
Хелатор Cu2+, тушитель HO•, LOO•,
HOCl |
|
Низкомолекулярные соединения |
||
|
Витамин Е |
Биомембраны |
Тушение HO•,
LOO•, HOCl и т. п. |
|
Убихинол |
Биомембраны |
Тушение HO•,
LOO•, HOCl и т. п. |
|
Каротиноиды |
Биомембраны |
Тушение HO•,
LOO•, HOCl, 1О2 |
|
Витамин С |
Цитоплазма |
Тушение HO•,
О2 |
|
Карнозин |
Цитоплазма |
Тушение HO•,
О2 |
|
N-ацетилцистеин |
Цитоплазма |
Неизбирательное тушение АФК |
|
Таурин Глутатион |
Цитоплазма Цитоплазма, митохондрии |
Нейтрализация
гипохлорита Тушение HO•,
О2 |
|
Мочевая кислота |
Кровь |
Предотвращение ПОЛ |
|
Билирубин |
Кровь |
Предотвращение ПОЛ |
В табл. 5
приведены концентрации низкомолекулярных антиоксидантов. Ясно, что они
значительно выше, чем АФК. Заметим, что среди низкомолекулярных антиоксидантов
важную роль играют пищевые вещества: витамины С и Е и
каротины. Такими же свойствами обладают ураты и
билирубин, которые ранее считали просто ненужными и даже вредными метаболитами.
Таблица 5. Концентрация
антиоксидантов в тканях, М
|
Вещество |
Печень |
Плазма |
|
Глутатион |
10–2 |
10–5 |
|
Аскорбат |
2∙10–3 |
5∙10–5 |
|
Ретинолы |
10–4 |
10–6 |
|
Токоферолы |
4∙10–6 |
2∙10–5 |
|
Ураты |
|
3∙10–4 |
|
Каротины |
|
3∙10–6 |
|
Билирубин |
|
10–5 |
Антиоксиданты водной фазы. Непосредственными предшественниками
гидроксильного радикала, инициирующего цепное окисление липидов, окислительную
модификацию белков и нуклеиновых кислот, служат ионы двухвалентного железа и
перекись водорода (или образующийся из нее гипохлорит). По этой причине
образование радикала гидроксила и свободнорадикальное
окисление биомолекул тормозятся веществами,
снижающими концентрацию одного из этих двух соединений. К ним относятся:
1. Фермент супероксиддисмутаза, который снижает концентрацию супероксидных радикалов и тем самым препятствует
восстановлению ими ионов трехвалентного железа до
двухвалентного. В клетке ионы железа хранятся в трехвалентном состоянии в
специальных депо, образованных субъединицами белка ферритина.
2. Ферменты каталаза и глутатионпероксидаза,
которые удаляют перекись водорода. Эффективность работы глутатионпероксидазы
зависит от концентрации свободного глутатиона,
при снижении которой может возрастать концентрация цитотоксических
гидроксильных радикалов. Регенерация восстановленного глутатиона (GSH) из окисленного (GSSG) осуществляется за
счёт НАДФН; этот процесс катализируется ферментом глутатионредуктазой. Недостаток глутатиона в клетках, например эритроцитах, который может
быть обусловлен действием токсических веществ, например, ионами тяжелых
металлов или наследственным недостатком глутатионредуктазы, приводит к
активации перекисного окисления; это, в частности, наблюдается при некоторых
видах гемолитических анемий.
Антиоксиданты, тормозящие развитие
цепных реакций в липидной фазе. Цепные реакции «ведут» свободные радикалы липидов (L•
и LOO•), разветвление цепей происходит при взаимодействии продукта пероксидации – гидроперекиси липидов (LOOH) с ионами Fe2+.
Все соединения, снижающие концентрацию перечисленных веществ, выполняют функцию
антиоксидантов. Сюда относятся:
1. Ферменты фосфолипаза и глутатионпероксидаза,
которые разрушают гидроперекиси липидов, предотвращая разветвление цепей
окисления липидов в мембранах. При этом действие фосфолипазы заключается в отщеплении от фосфолипидов
окисленной жирной кислоты, содержащей гидроперекисную
группу (LOOH), а действие глутатионпероксидазы
сводится к восстановлению этой группы до спиртовой (рис. 11) с одновременным
окислением глутатиона (GSH) до дисульфида (GSSG):
LOOH + 2GSH → LOH + GSSG +
H2O.
2. Ловушки
радикалов, которые называют иногда «липидными антиоксидантами». По своей
химической природе липидные антиоксиданты – это производные фенола. К ним
относится α-токоферол
(витамин Е), убихинон (коэнзим
Q), тироксин и синтетические соединения, например ионол
(бутилированный гидрокситолуол).

Рис. 11. Свободнорадикальная активация процессов перекисного окисления
липидов. Радикал Х* атакует ненасыщенные связи жирных кислот, приводя к
образованию липидных радикалов (а). Процесс ПОЛ, инициируемый
этими радикалами, осуществляется как цепная реакция, которая приводит к
накоплению различных липоперекисей (б). Последние
вызывают нарушения упаковки мембраны и внедрение в области мембранных дефектов
молекул воды и гидрофильных соединений, в т. ч. ионов Са2+.
Кальций активирует фосфолипазу (Фл),
которая расщепляет дефектную молекулу липида. Легче всего окисляются фосфолипиды,
содержащие полиненасыщенную арахидоновую
кислоту. Высвобождение арахидоновой кислоты позволяет
использовать ее для образования биологически активных соединений. В дальнейшем
специальный фермент глутатионпероксиидаза (Глу-Пер) обеспечивает репарацию мембраны (Болдырев, 2001)
3.
Соединения, связывающие железо. Большинство из них, включая такие природные
соединения как дипептид карнозин, не просто связывают
железо, но, самое главное, не дают ему возможности проникнуть в липидную фазу
мембран, поскольку образующиеся комплексы в силу своей полярности не проникают
в гидрофобную зону.
Для детоксикации двухвалентного железа в
организме существует, по-видимому, целая система окисления и связывания ионов
железа. В плазме крови эта система представлена ферментом церрулоплазмином (феррооксидазой),
который окисляет Fe2+ до Fe3+ кислородом без образования
свободных радикалов, и белком трансферрином, который
связывает и переносит в кровяном русле ионы трехвалентного железа, которые
затем захватываются клетками. В клетках железо может восстанавливаться
аскорбиновой кислотой и другими восстановителями, но затем окисляется и
депонируется в окисленной форме внутри ферментного белкового комплекса ферритина.
6.2. Краткая характеристика отдельных компонентов системы антиоксидантной
защиты организма
Токоферол
– основной жирорастворимый
антиоксидант организма:
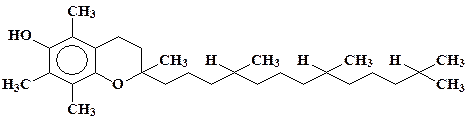
α-Токоферол,
или витамин Е, является по сути дела единственным и
самым мощным липидорастворимым антиоксидантом как в
плазме, так и в любой клеточной мембране.
α-Токоферол
способен прервать цепную реакцию за счет образования стабильного радикала.
Отдавая лабильный атом водорода липидному пероксильному
радикалу, α-токоферол превращается в радикал.
Образующийся радикал α-токоферола является
энергетически стабильным и обладает низкой реактивностью по отношению к другим
молекулам мембраны. Окисленный α-токоферол
восстанавливается в исходную форму при участии аскорбиновой кислоты (рис. 12).

Рис. 12. Цепные реакции витамина Е и С с липидными пероксильными радикалами.
Общепринято,
что действие α-токоферола сводится к следующим
механизмам:
1) защита от избыточного ПОЛ за счет очень высокой антирадикальной
активности. Подобно другим фенольным производным, α-токоферол
взаимодействует с радикалами как донор водорода и ловушка электронов, а его
углеводородный «хвост» является каналом удаления радикалов из углеводородной
зоны мембран;
2)
стабилизация липидного состава и физического состояния бислоя
(фактор структурной стабилизации мембран);
3) защита от
деструкции, вызванной продуктами гидролиза фосфолипидов под действием фосфолипазы А2: фосфолипаза
деполяризует мембрану, снижает ее микровязкость и
увеличивает ее отрицательный поверхностный потенциал за счет образования
свободных жирных кислот и фосфатидной кислоты, в то
время как α-токоферол связывает продукты
гидролиза фосфолипидов и уменьшает хаотропный эффект.
Кроме того, α-токоферол повышает микровязкость мембран, тем самым снижая пассивную
проницаемость для ионов;
4)
блокирование повреждающего действия синглетного
кислорода и других активных форм кислорода.
При
недостатке α-токоферола может произойти
разобщение тканевого дыхания и окислительного фосфорилирования,
уменьшается поглощение кислорода, концентрация убихинона,
содержание ферментов дыхательной цепи.
Имеются
данные о том, что около 20 % α-токоферола в плазме переносится с фракцией
липопротеинов низкой плотности (ЛПНП), а еще 50 % – с фракцией липопротеинов
высокой плотности (ЛПВП), что определяет антиоксидантные свойства последней.
Кроме того, исследователи выделяют в плазме особый белок, связывающий и
переносящий α-токоферол. Между ЛПВП и ЛПНП
происходит обмен α-токоферола. Кроме того,
доказано, что существует обмен α-токоферола между
плазмой и эритроцитами: этот обмен занимает 84–86 ч и находится в зависимости
от уровней гематокрита и общей концентрации липидов в плазме.
Таким
образом, витамин Е встраивается в биологические
мембраны и структурирует их подобно холестерину. Однако, в отличие от
последнего, α-токоферол преимущественно
включается в участки с наибольшим содержанием неэстерифицированных жирных
кислот (в частности, со стороны цитоплазмы и во внутренней мембране
митохондрий). Гидрофильное кольцо обращено к поверхности мембраны, а
гидрофобный «хвост» – внутрь мембраны, обеспечивая максимальное физическое
взаимодействие с неэстерифицированными жирными кислотами, в первую очередь с
длиной углеводородного радикала 16–20 атомов, т. е. сопоставимую с длиной
углеводородного «фитиля» α-токоферола. Следует
помнить, что это не просто «заполнение пустот», а специфическое межмолекулярное
взаимодействие. Одна молекула α-токоферола
реагирует более, чем с одной молекулой фосфолипидов, т. к. боковые фетильные радикалы размещаются в карманах, где имеется цис-двойная связь неэстерифицированных жирных кислот.
Именно такая локализация молекулы α-токоферола в биомембранах
и частицах фосфолипидов обеспечивает антиоксидантные свойства витамина Е и его способность быть стабилизатором мембран.
Убихинон (коэнзим Q) является обязательным и наиболее подвижным
компонентом электрон-транспортных цепей:
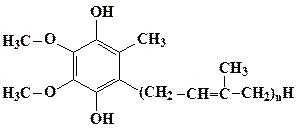
Убихинон
участвует в удалении протонов из матрикса митохондрий и последующем
освобождении их в межмембранное пространство. В соответствии с общепринятой в
настоящее время хемиосмотической моделью Питера
Митчелла это обеспечивает сопряжение процессов электронного транспорта и
окислительного фосфорилирования. Кроме того,
восстановленная форма убихинона, благодаря своей
способности присоединять электроны, служит хорошим антиоксидантом.
Антиоксидантная
функция убихинона была доказана после того, как
снижение содержания убихинона в митохондриях
сопровождалось усилением перекисного окисления, а его восстановление – обратным
эффектом. Восстановленный убихинон является
единственным липидорастворимым антиоксидантом,
который синтезируется в клетках животных и человека, а также постоянно
регенерируется из окисленной формы с помощью ферментных систем организма. Обе
формы убихинона встречаются во всех клеточных
мембранах, в плазме крови и липопротеинах низкой плотности.
Метаболизм убихинона
тесно связан с метаболизмом другого липофильного
антиоксиданта – витамина Е, являющегося наиболее
эффективным антиоксидантом в миокарде. Убихинон способен регенерировать восстановленную форму витамина Е. Их
концентрации в плазме пропорциональны содержанию липопротеинов, в частности оба
антиоксиданта прямо коррелируют с уровнем холестерина. При этом концентрация
витамина Е в плазме в несколько раз выше, чем убихинона, а в тканях – ситуация обратная.
Витамин С. Витамин С,
взаимодействуя с токоферолом и глутатионом,
является одним из ведущих компонентов биологической антиоксидантной системы.
Аскорбиновая кислота служит донором водорода для фермента аскорбат-пероксидазы,
разрушающей перекиси. Доказано стимулирующее влияние витамина
С на активность цитохрома Р450 – ключевого
фермента гидроксилирования и перекисного окисления.
Витамин С в форме аскорбат-иона –
наиболее важный эндогенный антиоксидант плазмы крови, он защищает липиды от
окисления пероксидными радикалами. Витамин С проявляет свойства антиоксиданта особенно в гидрофильной
среде. Высокие концентрации аскорбата определяются в
хрусталике, роговице, почках, головном мозге, поджелудочной железе, сердце.
Ткани этих органов метаболически очень активны, в
больших количествах потребляют кислород и нуждаются в аскорбате
для ферментных реакций и для защиты от окислителей. Антиоксидантный эффект аскорбата проявляется при достаточном количестве и активности
в организме других антиоксидантов, в частности токоферола, глутатиона.
При их недостатке может превалировать прооксидантный
эффект аскорбата и его метаболитов. Прооксидантные свойства могут проявляться и при избыточном
содержании аскорбиновой кислоты и, особенно, ее окисляющихся метаболитов – монодегидроаскорбата и дегидроаскорбата.
В присутствии меди и железа аскорбиновая кислота способствует образованию
перекиси водорода и супероксидного иона.
Fe3+ + Вит C → Fe2+ + Вит C•+ 2H+
2Вит C•+ 2H+ → Вит C + ДАК (дегидроаскорбат)
Вит C + Fe3+ → ДАК + Fe2+
Вит C + O2 → ДАК + H2O2
Таким
образом, аскорбиновая кислота обладает не только антиоксидантными, но и прооксидантными свойствами.
Антиоксиданты, содержащие в своей структуре
восстановленную сульфгидрильную группу или нуждающиеся в восстановленном
тиоле для проявления биологической активности. В составе
водорастворимых антиоксидантов ключевое место принадлежит веществам, содержащим
восстановленную сульфгидрильную группу (SH-группу) или нуждающихся в присутствии
тиолов для проявления активности. Так,
в состав неферментативного звена АОС входят
низкомолекулярные тиолы (восстановленный глутатион) и тиолсодержащие
белки, которые по некоторым данным даже более реактивны
по отношению к свободным радикалам, чем восстановленный глутатион
(к такого рода белкам относится, например, альбумин). С другой стороны,
ферменты, принимающие участие в противоокислительной
защите, либо являются собственно тиоловыми энзимами,
либо нуждаются в присутствии тиолов (СОД, каталаза,
ГПО). Исключительно высокая реакционная способность этих соединений делает
возможным их участие в самых разнообразных химических превращениях, но самую
важную в биологическом смысле роль играют окислительно-восстановительные
реакции, в ходе которых тиоловые группы легко
окисляются с образованием, как правило, дисульфидных
группировок, и вновь регенерируют при их восстановительном расщеплении: 2R–SH =
R–S–S–R + 2Н+.
Следует
помнить, что восстановленные тиолы обладают высокой
антиокислительной активностью, они имеют как антирадикальные,
так и антиперекисные свойства, и способны защищать от
повреждения ферменты и нуклеиновые кислоты, липиды и другие
биологически активные соединения.
Ключевое
место среди тиоловых антиоксидантов небелковой
природы занимает трипептид глутатион
(рис. 13), участвующий в обезвреживании различных АФК.

Рис. 13. Струкутура
глутатиона: 1 – остаток глутаминовой
кислоты; 2 – остаток цистеина; 3 – остаток глицина
В АОС роль глутатиона (GSH) заключается в том,
что: 1) это главный восстановитель клетки, его концентрация (1–10 мМ) выше, чем большинства органических веществ; 2) как и
другие низкомолекулярные антиоксиданты, он прямо восстанавливает АФК; 3)
функционирует на трех линиях ферментативной защиты (восстановление Н2О2,
ROOH и обезвреживание вторичных метаболитов окислительной модификации) из
четырех; 4)
GSH-зависимые ферменты работают во всех частях клетки, включая ядро,
митохондрии и эндоплазматическую сеть. Известный антиоксидатный
эффект Se также в основном опосредован ферментами –
обеими ГПО. Особое значение это вещество
играет в жизнедеятельности эритроцитов
и лимфоцитов, обеспечивая в первом случае защиту от окисления гемоглобина, а во втором – пролиферацию, продукцию
иммуноглобулинов и синтез цитокинов.
Следует
отметить, что тиоловое звено системы АОЗ занимает
особое место: между суммарной антиокислительной активностью и уровнем восстановленных тиолов нет
линейной зависимости, индивидуальный уровень тиолов
более стабилен, нежели общая антиокислительная активность. Это говорит о том,
что в приспособительных реакциях организма участвуют два фланга АОЗ, но
отражают они разные стороны клеточного метаболизма.
Антиоксидантные свойства белков
плазмы крови. Говоря
о факторах системы АОЗ, нельзя не затронуть вопрос об антиоксидантных свойствах
плазменных белков. Белки плазмы крови могут инактивировать активные формы
кислорода, а также связывать ионы переменной валентности, инициирующие
образование АФК, что позволило даже сформулировать представление об
«антиоксидантной белковой буферной системе», оказывающей в первую очередь
защиту на уровне эритроцитов, предотвращая их гемолиз в результате активации
ПОЛ.
Проблема заключается в том, что во внеклеточной среде активность
антиокислительных защитных ферментов (ГПО, каталазы, СОД) мала, но тем не менее
плазма обладает мощным антиокислительным потенциалом, который проявляют
альбумин, иммуноглобулины, церулоплазмин, фракции
α2- и β-глобулинов и в меньшей степени трансферрин, гаптоглобин и
сывороточная СОД. В предельно низких концентрациях эти белки практически не
влияют на скорость протекания реакций ПОЛ, но в средних концентрациях, которые,
однако, не достигают физиологических, они добиваются полной защиты эритроцитов
и легко окисляемых компонентов плазмы от окисления, проявляя при этом
выраженный кооперативный эффект.
Ключевое место среди белков плазмы принадлежит альбумину, который несет
основную антиокислительную функцию в плазме крови. Этот белок, кроме выполнения
роли основного осмотического компонента плазмы, выполняет транспортную функцию,
способен ассоциировать с разными лигандами
и влиять
на перенос их через мембраны. Среди веществ, транспортируемых альбумином,
ведущее место принадлежит билирубину, ионам кальция, различным лекарственным
препаратам и, конечно же, жирным кислотам, для которых в молекуле альбумина
имеются специфические независимые центры с высокой избирательностью,
недоступные для других лигандов. Обратимое связывание
альбумином и другими белками крови биологически активных веществ очень
тесно связано с нативным состоянием восстановленных тиоловых и дисульфидных
группировок на поверхности молекул белка и в центрах связывания.
Связывая
жирные кислоты, в первую очередь неэстерифицированные жирные, альбумин
предохраняет их от пероксидации. С другой стороны,
альбумин способен связывать и тем самым инактивировать продукты их окисления,
таким образом защищая клеточные структуры от
повреждающего действия продуктов ПОЛ при патологии.
Следует
помнить, что при чрезмерной активации ПОЛ окислительной модификации
подвергаются также и белковые компоненты АОС, что приводит к
потери ими антиокислительных свойств.
Литература
1.
Барабой В.А. Биоантиоксиданты. – Киев: Книга плюс, 2006. – 462 с.
2.
Болдырев А.А. Окислительный стресс и мозг // Соросовский образоват. журн. –
2001. – Т. 7, № 4. – С. 21–28.
3.
Костюк В.А., Потапович А.И. Биорадикалы
и биоантиоксиданты. – Мн.: БГУ, 2004. – 179 с.
4.
Кулинский В.И., Колесниченко Л.С. Биологическая роль глутатиона //
Успехи совр. биол. – 1990. – Т. 110, вып. 4. – С. 20–33.
5.
Меньшикова Е.Б., Зенков
Н.К. Антиоксиданты и
ингибиторы радикальных окислительных процессов // Успехи совр.
биологии. – 1993. – № 4. – С. 112–117.
6.
Brigelius-Flohe
R., Traber M.G. Vitamin E:
function and metabolism // FASEB J. – 1999. – V. 13. – P. 1145–1155.
7.
Halliwell
B., Aeschbach R., Loliger
J., Aruoma O.I. The
characterization of antioxidants // Food Chem. Toxicol.
– 1995. – V. 33 – P. 601–617.
8.
Linster C
L., Van Schaftingen E. Vitamin C.
Biosynthesis, recycling and degradation in mammals // FEBS Journal. – 2007. –
V. 274. – P. 1–22.
9.
Sies
H. Glutathione and its role in cellular functions //
Free Radic. Biol. Med. – 1999. – V. 27. – P. 916–921.
10.
Rice
M.E. Ascorbate regulation and
its neuroprotective role in the brain // Trends Neurosci. – 2000. – V. 23. – P. 209–216.
6.3. Определение общей
антиокислительной активности
6.3.1. Определение общей антиокислительной активности плазмы крови
Принцип
метода. Ненасыщенные жирные кислоты подвергаются перекисному окислению в
присутствии ионов двухвалентного железа. Об
интенсивности перекисного окисления судят по накоплению в среде инкубации МДА.
По степени торможения накопления МДА в присутствии плазмы крови судят о ее суммарной АОА.
Оборудование. Центрифуга с
охлаждением, термостат, электроплитка, фотоэлектроколориметр
типа КФК-3 или спектрофотометр СФ-46, магнитная мешалка.
Реактивы и
их приготовление.
1. Суспензия линоленовой кислоты. К 1 мл дистиллированной воды (37 °С)
добавляют 50 мкл тритона Х-100, после перемешивания
добавляют 10 мкл линоленовой кислоты. Хорошо
перемешивают и переносят в стаканчик с 24 мл дистиллированной воды.
Суспензию постоянно перемешивают на магнитной мешалке. Готовят каждый день.
2. 1 мМ раствор сульфата железа (II). 27,8 мг соли растворяют в 100 мл
дистиллированной воды.
3. 0,8 %-ный раствор тиобарбитуровой
кислоты. Объем приготовляемого раствора ТБК зависит от количества проб. 80 мг
ТБК при нагревании (60–70 °С) растворяют в 10 мл дистиллированной воды. Раствор
готовят перед опытом.
4. 0,9 %-ный раствор хлорида натрия.
5. 1 %-ный раствор фосфорной кислоты. 3,4 мл
концентрированной кислоты (плотность 1,705 г/мл) разбавляют дистиллированной
водой до 500 мл.
6. Бутанол.
Ход
определения. Отобранную плазму крови разводят, для чего к 0,1 мл
плазмы добавляют 0,3 мл физиологического раствора.
Для определения общей АОА используют две пробы: контрольную (без плазмы
крови) и опытную (с плазмой крови). С этой
целью в 2 пробирки помещают 0,5 мл суспензии линоленовой кислоты, 0,1 мл 1 мМ раствора сульфата железа. В опытную пробу добавляют 0,01
мл разведенной плазмы крови, а в контрольную – 0,01 мл физиологического
раствора. Параллельно ставят контрольные пробы, где вместо линоленовой кислоты
берут 0,5 мл дистиллированной воды. Все пробы инкубируют в течение 1 ч при 37 °С, постоянно перемешивая.
Накопление в среде инкубации МДА устанавливают с помощью ТБК (Андреева и
др., 1988). Для этого по окончании инкубации из каждой пробы отбирают 0,2 мл,
добавляют 3 мл 1 %-ной фосфорной кислоты и после перемешивания добавляют 1 мл
0,8 % раствора ТБК. Пробы хорошо перемешивают и ставят в кипящую водяную баню
на 1 ч, затем охлаждают до комнатной температуры. В каждую пробирку добавляют 4
мл бутанола, тщательно перемешивают и для разделения фаз центрифугируют 10 мин
при 3000 об./мин. Измеряют
оптическую плотность верхней бутанольной фазы при
длине волны 515 и 532 нм против контрольной пробы. В
расчет берут разность экстинкций: DЕ = Е532 – Е515.
АОА вычисляют в процентах по формуле, где за 100 % принимается активность
такого антиоксиданта, который полностью подавляет образование МДА в опытной
пробе за 1 ч инкубации:
 ,
,
где [МДАоп]
– содержание МДА (в единицах экстинкции) в опытной пробе с добавкой плазмы
крови; [МДАк]
– содержание МДА в контроле без биоматериала соответственно в начальный момент
времени (0¢) и после инкубации (60¢).
6.3.2. Определение общей
антиокислительной активности гомогената головного
мозга
Определение
общей антиокислительной активности гомогената мозга
производится так же, как в плазме крови, в соответствии с методом М.Л. Демчук и др. (1990).
Различие
заключается в следующем. Из мозга готовят 10 %-ный гомогенат на 0,9 %-ном растворе хлорида натрия. Гомогенат используется без разбавления. В опытную пробу
добавляют 0,1 мл 10 %-ного гомогената,
а в контрольную – 0,1 мл дистиллированной воды. Антиокислительную активность
вычисляют в процентах, как для плазмы крови.
Литература
1. Андреева Л.И., Кожемякин А.А., Кишкун А.А. Модификация метода определения
перекисей липидов в тесте с тиобарбиталовой кислотой
// Лаб. дело. – 1988. – № 11. – С. 41–43.
2. Демчук
М.Л., Левченко Л.И., Промыслов М.Ш. Процессы перекисного окисления липидов при черепно-мозговой
травме // Нейрохимия. – 1990. – Т. 9. – № 1. – С.
108–110.
6.4. Определение антиокислительной
активности
6.4.1. Определение антиокислительной активности гидрофильных компонентов плазмы крови
Принцип метода. Метод основан на регистрации скорости окисления
восстановленной формы 2,6-дихлорфенолиндофенола (2,6-ДХФИФ) кислородом,
растворенным в реакционной среде при наличии и отсутствии биологического
материала. При этом бесцветная лейкоформа
2,6-ДХФИФ переходит в окрашенную форму, имеющую максимум поглощения при 600 нм. Константа ингибирования биологическим материалом
окисления 2,6-ДХФИФ служит показателем антиокислительной активности
биологического материала. Выбор данного субстрата обусловлен тем, что его окислительно-восстановительный потенциал (0,22 В при pH 7,9) позволяет быстро и количественно переводить его из
окисленной формы в восстановленную в присутствии сульфата двухвалентного
железа. Кроме того, 2,6-ДХФИФ способен окисляться
кислородом воздуха с удобной для регистрации скоростью в стационарных условиях.
Реактивы и их приготовление.
1.
2. 0,8 мМ
водный раствор 2,6-ДХФИФ в окисленной форме. Для этого 11,6 мг
2,6-дихлорфенолиндофенола растворяют в 50 мл дистиллированной воды. Раствор
готовят непосредственно перед анализами.
3. 3,2 мМ
раствор закисного сернокислого железа. 44,5 г FeSO4×7Н2O растворяют в 50 мл дистиллированной
воды. Раствор готовят непосредственно перед анализами.
Оборудование. Cпектрофотометр с термостатируемой
кюветой или спектрофотометр «Спекол» с термостатируемой кюветой; ультратермостат;
баня водяная; весы аналитические; колбы мерные; пипетки измерительные;
микропипетки на 10 и 20 мкл.
Ход определения.
В термостатируемую кювету последовательно добавляют
0,5 мл фосфатного буфера (рН 7,4), 0,15 мл раствора 2,6-дихлорфенолиндофенола,
0,15 мл раствора закисного сернокислого железа и быстро перемешивают. Все
растворы должны иметь температуру 37 ºС. Сразу после перемешивания быстро
добавляют 10 или 20 мкл плазмы крови (опыт) или воды
(контроль), перемешивают и через каждые 30 с после добавления плазмы или воды
на протяжении 5 мин измеряют оптическую плотность при 600 нм
против воды (Don, Dк).
Определяют оптическую плотность при 600
нм инкубационной среды, в которой 2,6-ДХФИФ окислен
полностью. Для этого в кювету добавляют те же компоненты, но вместо раствора FеSO4 и пробы добавляют равные им объемы
дистиллированной воды (Dmax).
Расчет результатов начинают с
определения констант скоростей окисления 2,6-ДХФИФ в контроле (Кк) и
опыте (Коп) по формулам:
![]() и
и ![]() ,
,
где ln – логарифм натуральный
(положительный).
Рассчитывают константу ингибирования (Ки)
плазмой крови окисления 2,6-ДХФИФ, являющуюся показателем ее
антиокислительной активности по формуле
![]() ,
,
где Ки
– антиокислительная активность плазмы крови, мл·мин–1;
Кк – константа скорости
окисления 2,6-ДХФИФ в контроле; Коп
– константа скорости окисления 2,6-ДХФИФ в опыте; С – концентрация
плазмы в инкубационной смеси, мг/л.
Примечания: 1) плазма крови должна быть без гемолиза; 2)
исследование должно проводиться не позднее 6 ч после взятия крови.
6.4.2. Определение антиокислительной активности гидрофильных
компонентов головного мозга
Определение
антиокислительной активности гидрофильных компонентов головного мозга
производят так же, как и плазмы крови, в соответствии с методом В.Л. Семенова и
А.М. Ярош (1985). Различия заключаются в следующем.
Из выбранного отдела головного мозга готовят 10 %-ный
гомогенат на 0,9 %-ном растворе хлорида натрия. Гомогенат центрифугируют при 3000 об./мин в течение 10 мин. Супернатант
отбирают и разводят 0,9 %-ным раствором хлорида
натрия в соотношении 1:10 и используют для анализа.
Константу
ингибирования (Ки) гомогенатом
мозга окисления субстрата вычисляют из следующего выражения:
![]() ,
,
где Ки – антиокислительная
активность гомогената мозга, мг мин–1;
Кк – константа скорости
окисления 2,6-ДХФИФ в контроле; Коп
– константа скорости окисления 2,6-ДХФИФ в опыте; С – концентрация
ткани мозга в инкубационной смеси, мг/л.
Литература
Семенов В.Л., Ярош А.М. Метод определения антиокислительной
активности биологического материала // Укр. биохим. журнал. – 1985. – Т. 57, № 3. – С. 50–52.
6.5. Определение
антиокислительной активности сыворотки крови по Глевинду
Принцип метода. Для определения антирадикальной
активности используют раствор устойчивого свободного радикала –
1,1-дифенил-2-пикрилгидразила, который восстанавливается в реакции с
антиоксидантом, при этом оптическая плотность его раствора, измеряемая при
длине волны 517 нм, резко снижается.
Реактивы и их приготовление.
1. Толуол для
спектроскопии.
2. 10 % раствор
фосфорно-вольфрамовой кислоты.
3. Раствор
1,1-дифенил-2-пикрилгидразила в метаноле с оптической плотностью 0,7–0,73.
4.
Аскорбиновая кислота, раствор 0,1 мкэкв в 1 мл.
Ход определения. Для определения содержания
антиоксидантов в сыворотке крови белки осаждают фосфорно-вольфрамовой кислотой.
Для этого к 0,25 мл сыворотки добавляют 0,25 мл раствора фосфорно-вольфрамовой
кислоты. Объем фильтрата доводят до 1,5 мл бидистиллированной
водой и добавляют 3 мл раствора 1,1-дифенил-2-пикрилгидразила в метаноле. Пробы
хорошо перемешивают, через 5 мин добавляют 4 мл толуола и встряхивают. Для
разделения фаз смесь центрифугируют при 1000 об./мин в течение 10 мин. Оптическую
плотность верхнего окрашенного слоя измеряют при длине волны 517 нм против толуола. Контрольная проба содержит 1,5 мл воды.
Для расчета используют разницу оптических плотностей контрольной
и опытных проб. Количество антиоксидантов определяют по калибровочной кривой,
для построения которой используют раствор аскорбиновой кислоты.
Результаты выражают в мкМ.
Литература
Glavind J.
Antioxidants in animal tissue // Acta Chem. Scand.
– 1963 – Vol. 17, № 13. – P. 1635–1640.
6.6. Определение содержания сульфгидрильных групп и дисульфидных
связей
в белках и низкомолекулярных соединениях
Среди
функциональных групп биологической системы SH-группам принадлежит важная роль в
поддержании структурной целостности и регуляции функциональной активности
клеточных компонентов. Их высокая реакционная способность обусловливает
активное взаимодействие с различными ацилирующими, алкилирующими, арилирующими и метилирующими веществами, галоидкислотами
и их амидами, дисульфидами и карбонильными соединениями, металлами, а также
активированными двойными связями, что имеет важное биологическое значение. Так,
благодаря этим свойствам тиолы принимают
непосредственное участие в регуляции митотического деления клетки, активности
ферментных комплексов, катализирующих образование наиболее важных
физиологически активных веществ, мышечном сокращении, свертывании крови,
проницаемости клеточных и субклеточных мембран, в поддержании структурой
целостности и функциональной активности молекулярных компонентов иммунной
системы.
SH-соединения как низкомолекулярной, так и белковой природы играют особую
роль в функционировании АОС. Проявляя антирадикальное и антиперекисное действие, тиолы
подвергаются обратимому (как правило) окислению с образованием соответствующих
дисульфидов, поэтому концентрация соединений, содержащих тиоловые
(-SH) и дисульфидные (-S-S-) функциональные группы, а также их
соотношение (тиолдисульфидный коэффициент SH/SS) могут служить количественной
характеристикой состояния неферментативного звена
АОС, представленного тиолами.
В белках по реакционной способности
различают три типа SH-групп: легко реагирующие, вяло реагирующие и
замаскированные. Однако в тканевых экстрактах различными методами выявляются
гораздо больше типов SH-групп, и часто им приписывают неодинаковые наименования.
По способу обнаружения методом
амперометрического титрования в тканевых экстрактах дифференцируются
легкодоступные, свободные, поверхностно расположенные и структурно
замаскированные белковые, а также суммарные SH-группы. Легкодоступные
SH-группы
представляют собой сумму свободных и поверхностно расположенных белковых SH-групп. После осаждения белков в надосадочной жидкости определяются свободные SH группы. Показано, что восстановленный глутатион
составляет основную массу свободных SH-групп. По разности между
легкодоступными и свободными SH-группами находят количество поверхностно расположенных
белковых SH-групп, которые в литературе встречаются под названиями белковые, белковосвязанные SH-группы. Структурно замаскированные белковые SH-группы представляют собой в основном
стерически спрятанные и химически связанные SH-группы растворимых белков. В
условиях амперометрического титрования они недоступны в таком виде к
определению. Естественно, что между различными типами SH-групп не существует резких граней и
не всегда легко отнести ту или иную группу к определенному типу. Тем не менее деление SH-групп на типы по реакционной способности полезно и мы в
своих исследованиях придерживались указанной классификации.
6.6.1. Амперометрический метод
определения низкомолекулярных и белковых сульфгидрильных групп
в крови
Для определения различных типов SH-групп в крови, белках мембран
эритроцитов и синаптосом использовали
амперометрический метод титрования. Этот метод является одним из видов
электрометрического объемного анализа. В своих исследованиях мы использовали
методику, разработанную профессором В.В. Соколовским на аппарате,
сконструированном в лаборатории кафедры биохимии Дагестанского государственного
университета. Схема установки для амперометрического титрования показана на
рис. 14.
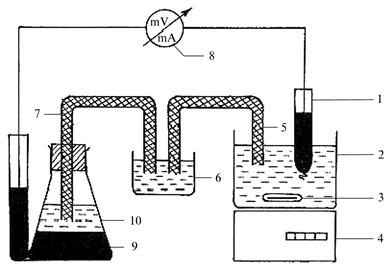
Рис. 14. Схема установки для
амперометрического титрования сульфгидрильных групп: 1 – платиновый электрод; 2
– ячейка с исследуемой пробой; 3 – остеклованный цилиндрик; 4 – магнитная
мешалка; 5 – электрический мостик (с нитратом аммония); 6 – ячейка с насыщенным
раствором нитрата аммония; 7 – электрический мостик (с хлоридом калия); 8 –
миллиамперметр; 9 – электрод сравнения с ртутью; 10 – электролит
Принцип метода. При титровании растворов, содержащих тиоловые
соединения, азотнокислым серебром ионы серебра связываются с SH-группами с образованием меркаптида по уравнению
R-SH
+ Ag+ → R-SAg + H+,
где R-SH – соединения, содержащие
сульфгидрильные группы; R-SAg – меркаптиды
серебра. По достижении конечной точки титрования (когда все SH-группы оказываются блокированными) в
растворе появляется избыток ионов серебра. При этом в
электрическом элементе, состоящем из погруженных в титруемый раствор
платинового индикаторного электрода и электрода сравнения, возникает
диффузионный ток, пропорциональный концентрации ионов серебра и измеряемый с
помощью миллиамперметра. Содержание SH-групп в исследуемом растворе
эквивалентно количеству раствора азотнокислого серебра, затраченного на
титрование. Чувствительность метода составляет 0,1–1,0 мкмоль
SH-групп.
Данным
методом определяются свободные и вяло реагирующие
сульфгидрильные группы нативных белков.
Замаскированные сульфгидрильные группы белков определяются только после их
денатурации.
Оборудование.
1. Милливольтмиллиамперметр М-198/1 с ценой
деления 10–8А или вольтамперметр универсальный В7-21А, В7-4Б и др.
2. Платиновый
индикаторный электрод. Это стеклянная трубка длиной 100–150 мм, в нижний конец
которой впаяна платиновая проволочка так, чтобы снаружи находилась часть
проволоки длиной 5–8 мм. Трубку заполняют металлической ртутью, в которую
свободно погружен конец проводника (стальная проволока; d = 0,5 мм), связывающего электрод с амперметром.
3. Электрод
сравнения. Электродом служит слой ртути, залитый в специальный сосуд (рис. 14).
Над ртутью находится раствор электролита (йодид калия и йодид ртути в
насыщенном растворе хлорида калия). В раствор электролита опускают проводник,
связывающий электрод сравнения с амперметром, и один конец солевого мостика,
представляющего собой U-образную стеклянную трубку, заполненную агаровым гелем, приготовленным
на насыщенном растворе хлорида калия. Другой конец солевого мостика электрода
сравнения опускают в ячейку с насыщенным раствором нитрата аммония. В эту же
ячейку опускают вторую U-образную стеклянную трубку с агаровым гелем, приготовленным
на насыщенном растворе нитрата аммония. Другой конец этого солевого мостика
опускают в ячейку для титрования.
Реактивы и их приготовление.
1.
2.
3.
4. Насыщенный
раствор хлорида калия.
5. 30 %-ный раствор азотнокислого аммония.
6. Гель для
заполнения электрического мостика. При нагревании на водяной бане растворяют
7. Раствор
электролита для электрода сравнения.
8. Ртуть металлическая. Очистка ртути осуществляется
следующим образом: ртуть промывают разбавленной азотной кислотой (разведенной
дистиллированной водой в отношении 1:1), затем многократно промывают
дистиллированной водой и фильтруют через плотную ткань. Все работы с ртутью производят под тягой в приспособленном для этого
помещении.
9. 0,001 н.
водный раствор азотнокислого серебра.
10. 1 %-ный раствор додецилсульфата
натрия.
11. 6 %-ный раствор сульфосалициловой кислоты.
12. Реактивы
для определения белка по Лоури.
13. Растворы
для выделения мембран эритроцитов.
Ход определения. Плазма крови. Для
определения суммарного (содержания как в белках, так и низкомолекулярных
соединениях) количества SH-групп к 0,2 мл плазмы добавляют 1 мл 1 %-ного
раствора додецилсульфата натрия и выдерживают 5 мин
при 37 °С, периодически перемешивая. Затем общий объем
пробы доводят аммиачным буфером до 20 мл и титруют.
При
определении содержания тиоловых групп
низкомолекулярных соединений к 0,5 мл плазмы добавляют 0,7 мл дистиллированной
воды и 0,8 мл 6 %-ного раствора сульфосалициловой
кислоты. Через 10 мин белки осаждают центрифугированием при 4000 об./мин в течение 10 мин. Из надосадочной жидкости отбирают 1 мл и к нему добавляют 19
мл аммиачного буфера для титрования.
Гемолизат. К 0,5 мл
трижды отмытых эритроцитов добавляют 9,5 мл холодной бидистиллированной
воды и оставляют на 10 мин на холоде для гемолиза. Неразрушенные клетки и
стромы эритроцитов осаждают при 10000 об./мин в течение 20 мин. Для определения суммарного количества SH-групп из верхнего слоя жидкости (гемолизата) отбирают 0,1 мл и вносят в сосуд для
титрования, содержащий 19,9 мл аммиачного буфера.
Для
определения количества низкомолекулярных тиоловых
соединений к 1 мл гемолизата добавляют 0,8 мл 6 %-ного раствора сульфосалициловой кислоты. После осаждения
белков 1 мл надосадочной жидкости используют для
титрования.
Как в плазме,
так и в гемолизате количество белковых SH-групп рассчитывают по разности между
содержанием суммарных SH-групп и SH-групп низкомолекулярных тиоловых
соединений.
Мембраны эритроцитов. В белках
мембран эритроцитов общее количество свободных SH-групп складывается из поверхностных
(легкодоступных) и скрытых (труднодоступных).
При
определении суммарного количества SH-групп белков мембран эритроцитов 0,2
мл суспензии мембран солюбилизируют в 1 мл 1 %-ного раствора додецилсульфата
натрия (5 мин при 37 °С). Затем общий объем среды аммиачным буфером доводят до
20 мл и титруют.
Измерение
количества поверхностных SH-групп мембранных белков проводят в 0,4 мл исходной
суспензии.
По разности
между содержанием общих и поверхностных SH-групп рассчитывают количество
скрытых сульфгидрильных групп белков.
Порядок титрования. Для титрования собирают установку по схеме (рис. 14).
В нерабочем состоянии платиновый электрод погружен в стаканчик с
дистиллированной водой, конец агарового мостика электрода сравнения погружен в
насыщенный раствор азотнокислого аммония.
При анализе в
стаканчик с образцом и буфером опускают электрод, агаровый мостик,
остеклованный цилиндрик для перемешивания. Включают магнитную мешалку. После
того как показание измерительного прибора будет стабильным, начинают
титрование, добавляя по 0,05 мл 0,001 н. раствора азотнокислого серебра и
отмечая показания прибора.
Для
определения результатов титрования строят график, откладывая на оси ординат
показания величин тока, а на оси абсцисс – соответствующие им объемы
добавленного раствора азотнокислого серебра. Две прямые, соединяющие отмеченные
на графике точки, пересекаются в конечной точке титрования – точке
эквивалентности (рис. 15).
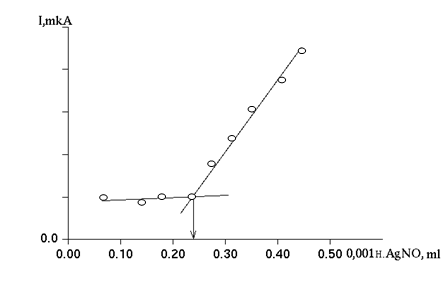
Рис. 15. Амперометрическое титрование
сульфгидрильных групп
Содержание SH-групп, эквивалентное количеству
затраченного на титрование раствора азотнокислого серебра, вычисляют, исходя из
расчета, что 1 мл 0,001 н. раствора азотнокислого серебра эквивалентен 1 мкмолю SH-групп (или 0,033 мг SH-групп).
Содержание
сульфгидрильных групп в плазме крови выражают в мкмолях на 100 мл, а в гемолизате
– в мкмолях на 100 мл упакованных эритроцитов:
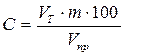
где С – количество SH-групп в мкмолях
на 100 мл; VТ – объем 0,001 н. раствора азотнокислого серебра, соответствующий
эквивалентной точке; m – разведение пробы; 100 –
коэффициент пересчета на 100 мл; Vпр – объем пробы, взятой на титрование.
Количество SH-групп в белках мембран эритроцитов
выражают в мкмолях SH-групп на 1 мг белка. Белок
определяют по методу Лоури.
Литература
1. Соколовский В.В. Амперометрический метод определения
низкомолекулярных и белковых сульфгидрильных групп // Вопр.
мед. химии. – 1977. – № 3. –
С. 15–20.
2. Торчинский Ю.М. Сера в белках. – М., 1977. – 302
с.
6.6.2. Количественное определение дисульфидных
связей
в белках и низкомолекулярных
соединениях крови
Принцип метода. Дисульфидные группы
восстанавливают сульфитом натрия: ![]()
Реакция
происходит в присутствии избытка ионов серебра, которые с новыми
SH-группами
образуют прочную меркаптидную связь, а избыток
свободных ионов серебра оттитровывают эквимолярным раствором унитиола.
Отбор проб и их хранение. Плазму крови, гемолизат
и мембраны эритроцитов получают по методике определения SH-групп.
Оборудование. Используют то же самое оборудование, что и для титрования SH-групп.
Реактивы и их приготовление. Кроме тех, что используют для
титрования SH-групп, необходимы следующие реактивы:
1) 5·10–4
М раствор унитиола. 0,21 мл 5 %-ного раствора ампулированного
препарата разбавляют дистиллированной водой до 100 мл (готовят ежедневно);
2) сульфит
натрия, кристаллический порошок.
Ход определения. В ячейку для титрования, содержащую 19 мл 0,01 М
аммиачного буфера (рН 9,2), последовательно вносят 0,5 мл 10–3 М
раствора азотнокислого серебра, 0,25 мл плазмы крови (0,1 мл 10 %-ного гемолизата; 0,2 мл суспензии
мембран эритроцитов, содержащей 3–4 мг белка на мл) и 2–3 мг сульфита натрия.
Смесь постоянно перемешивают на магнитной мешалке с заданной скоростью.
Обратное титрование проводят 5·10–4 М раствором унитиола,
добавляя его по 0,05 мл, после того, как показание регистрирующего прибора
будет стабильным. По точке эквивалентности вычисляют суммарное количество тиоловых групп (свободные SH-группы + SH-группы, образующиеся после
восстановления дисульфидных связей).
При
определении количества свободных SH-групп титрование биоматериала производят в тех же условиях,
но в отсутствие сульфита натрия. Содержание тиоловых
групп выражают в мкмолях на 100 мл плазмы или
упакованных эритроцитов (в случае гемолизата), а
также на мг белка (в случае мембран эритроцитов), используя следующие выражения:
плазма крови:
 ,
,
где С – концентрация тиоловых групп в мкмоль/100 мл
плазмы; DV – разность
между объемом азотнокислого серебра, содержащегося в пробе (0,5 мл), и объемом унитиола, израсходованного на титрование; 0,25 – количество
плазмы, использованное для анализа, мл;
гемолизат:
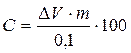
где m – разведение пробы при приготовлении гемолизата; 0,1 – количество гемолизата,
использованное для анализа, мл;
мембраны эритроцитов:
 ,
,
где К – количество
белка в пробе для анализа, мг.
Содержание дисульфидных связей рассчитывают по разности между
количеством SH-групп, обнаруженных в присутствии и отсутствие сульфита натрия.
Литература
Соколовский В.В., Белазерова Л.А.,
Огурцова Р.Е. Метод количественного определения дисульфидных
групп крови обратным амперометрическим титрованием // Лаб. дело. – 1977. – № 1.
– С. 26–28.
6.7. Определение содержания белковых и
низкомолекулярных тиолов спектрофотометрическим
методом
В антиоксидантной защите организма важное место принадлежит соотношению SH- и S-S-групп. Существуют различные подходы к изучению тиол-дисульфидного обмена. Один из таких подходов является
способ определения общих тиоловых групп и небелковых тиоловых групп с 5,5’-дитио-бис-(2-нитробензойной
кислотой), предложенный Эллманом (1959).
6.7.1. Определение суммарного
содержания тиоловых групп в сыворотке крови
Принцип метода. Метод основан на способности тиоловых
соединений при взаимодействии с 5,5’-дитио-бис-(2-нитробензойной
кислотой) (ДТНБ, реактив Эллмана) образовывать
окрашенное соединение – тио-2-нитробензойную кислоту, водный раствор которого
имеет максимум поглощения при λ = 412 нм.
Реактивы и их приготовление.
1.
2.
3. Реактив Эллмана: готовят маточный раствор путем растворения 39,6 мг
ДТНБ в 10 мл 10 мМ фосфатного буфера рН 7,0. Перед
определением реактив разводят в 150 раз 100 мМ
фосфатным буфером рН 8,0.
Ход определения. К 0,1 мл сыворотки крови добавляют
2,4 мл раствора ДТНБ (соотношение объемов 1:25). Через 30 мин инкубации при
комнатной температуре в темноте измеряют оптическую плотность проб при λ =
412 нм против контроля, содержащего вместо сыворотки
воду. Параллельно готовят холостую пробу: чтобы исключить мутность раствора,
0,1 мл сыворотки разводят 2,4 мл 0,1 М фосфатного буфера (рН 8,0) в соотношении
1:25.
Расчет
производят с учетом коэффициента молярной экстинкции 13×103 М–1см–1
и разведения сыворотки. Для расчета используют разницу оптических плотностей
опытной и холостой проб. Результаты выражают в мкмоль/л.
Литература
Соколовский В.В., Кузьмина В.С., Москадынова
Г.А., Петрова И.И. Спектрофотометрическое
определение тиолов в сыворотке крови // Клинич. лаб. диагностика. –
1997. – № 11. – С. 20–21.
6.7.2. Определение небелковых тиоловых групп в эритроцитах
Реактивы и их приготовление.
1. 20 %
раствор сульфосалициловой кислоты.
2.
3. Абсолютный
метанол.
4. 0,4 % метаноловый раствор ДТНБ (готовят перед определением).
Ход определения. К 0,6 мл гемолизата
эритроцитов (1 объем отмытой эритроцитарной взвеси и 10 объемов дистиллированной воды)
добавляют 0,2 мл 20 % раствора сульфосалициловой кислоты. Пробы перемешивают и
центрифугируют при 3000 об./мин
в течение 10 мин при температуре 2 °С. 0,2 мл супернатанта
переносят в пробирки, содержащие 2,55 мл трис-буфера
с ЭДТА. К полученной смеси добавляют 25 мкл раствора
ДТНБ. Измеряют оптическую плотность проб против дистиллированной воды при
λ = 412 нм в кювете с толщиной слоя 10 мм.
Расчёт производят с помощью калибровочной кривой, для построения которой
используют растворы восстановленного глутатиона с
концентрациями от 0,02 до 2,0 мМ.
Считается,
что около 90 % небелковых тиоловых групп эритроцитов
принадлежит восстановленному глутатиону,
поэтому приведенный здесь метод Эллмана может быть
использован для приблизительной оценки уровня глутатиона
в эритроцитах.
Литература
Медицинские
лабораторные технологии: справочник / под ред. А.И. Карпищенко.
– СПб.: Интермедика, 1999. –
Т. 2. – С. 72–73.
6.8. Определение содержания α-токоферола в
сыворотке крови
Принцип метода. Метод основан на измерении
флуоресценции α-токоферола, экстрагированного из
сыворотки крови гексаном.
Отбор проб и их хранение. Кровь из вены отбирают в сухую
пробирку или в пробирку, содержащую 0,1 мл гепарина. Отцентрифугированная
сыворотка или плазма может храниться в замороженном виде при – 20°С до 4 мес.
Оборудование. Спектрофлуориметр, центрифуга.
Реактивы и их приготовление.
1. Этиловый
спирт 96 %, перегнанный.
2. н-гексан, перегнанный в стеклянной
системе.
3.
α-токоферол, стандартный раствор 20 мкмоль/л в
этиловом спирте. Реактивы хранят в склянках темного стекла.
Ход определения. К 0,3 мл плазмы или сыворотки крови
добавляют 1 мл бидистиллированной воды и встряхивают
в течение 30 с. После этого добавляют 2 мл этилового спирта для осаждения
белков, снова встряхивают и оставляют стоять на 5 мин. Затем добавляют 3 мл н-гексана, энергично встряхивают в течение 1 мин и
центрифугируют при 1500 об./мин
в течение 2–3 мин. В верхнем гексановом слое измеряют интенсивность флуоресценции при длине волны
возбуждения 295 нм и длине волны испускания 323 нм.
В стандартную
пробу вместо сыворотки вносят 0,3 мл стандартного раствора α-токоферола,
в контрольную пробу – 0,3 мл этанола, дальнейшие процедуры проводят, как в
опытных пробах.
Концентрацию
экстрагированного из сыворотки крови α-токоферола
(С) в мкмоль/л рассчитывают по формуле
 ,
,
где Фо, Фк, Фс – интенсивность флуоресценции опытной
(сыворотка или плазма), контрольной и стандартной проб соответственно; Сс – концентрация α-токоферола
в рабочем стандартном растворе, мкмоль/л.
Литература
Методы исследований в профпатологии / под ред. О.Г. Архиповой. – М., 1988. – 207 с.